Past Seminars & Events
Tekalign Debela
Tekalign Debela
Department of Chemistry
University of Oregon
Abstract
Defects in earth-abundant layered materials for energy applications
Defects in crystalline structures play a central role in today’s technology, affecting the efficiency and reliability of devices in a variety of applications. These imperfections are particularly important for solar cells, batteries, thermoelectrics, catalysts, quantum computers and many others. It is therefore crucial to study the effects of structural defects on electronic properties in order to develop an application-specific defect engineering strategy. In my talk, I will discuss my computational work on modeling defects in two-dimensional (2D) materials, focusing on their role in improving the catalytic performance of transition metal dichalcogenides (TMDs) for the hydrogen evolution reaction (HER). This work, largely based on experimental collaboration, explores different types of defects, including: chalcogen vacancies, metal adatoms and non-stochiometric defects in TMDs. In addition, I will provide insights into my recent studies on hydrogenic defects in conductive 2D metal-organic frameworks (MOFs). MOFs are usually formed by connecting organic linkers with metal ions to form a three-dimensional network. During this process, defects can occur that have a significant impact on their properties. In particular, I will discuss how the hydrogen interstitials and vacancies, a process that is not easily observed in experiments, can be manipulated to change the properties of this material. These hydrogen-induced defects in MOFs are likely to be widespread and can be strategically utilized to alter the overall properties of these materials. This knowledge opens up new possibilities in materials science, particularly in the development of materials with tailored properties for specific applications.
Tekalign Debela
Dr. Tekalign T. Debela is currently a Postdoctoral Associate at the University of Oregon, working in the groups of Prof. Christopher H. Hendon and Prof. Shannon W. Boettcher in the Department of Chemistry and Biochemistry. His research focuses on the application of density functional theory to unravel the mechanisms of water dissociation at metal oxide/water interfaces and on the modeling of defects in metal-organic frameworks (MOFs). From 2017 to 2021, he was a research professor at Jeonju University, South Korea, where he focused on the computational design of layered materials for energy applications as part of the “Brain Pool” fellowship. Dr. Debela obtained his PhD in Materials Science and Engineering from Zhejiang University (China) in 2014. As part of his doctoral studies, he worked intensively on molecular dynamics simulations, particularly the study of nucleation and crystallization processes in metallic melts. His education began in Ethiopia, where he earned a BSc and MSc in Physics from Arba Minch University and Addis Ababa University, respectively.
Hosted by Professor Ilja Siepmann
Kevin Carter-Fenk
Kevin Carter-Fenk
University of California, Berkeley
Abstract
Exploiting the Butterfly Effect: How Choice Affects Change in Computational Spectroscopy
As spectroscopic technologies become more advanced, the exploration of photochemical processes in molecular systems provides increasingly intricate insights into the interplay of molecular interactions, elucidating the foundations of macroscopic structures, unraveling the mechanisms of charge transport crucial for designing solar energy materials, and even paving the way for innovative approaches to biological radiation therapies. Harnessing the predictive capabilities of quantum chemistry can provide a powerful toolkit to clarify and inform experimental results. However, common computational methods, such as time-dependent density functional theory (TDDFT), can exhibit dramatic errors that limit the utility of the calculated spectra. These errors are particularly pronounced for electronic excitations that significantly alter the charge distribution such as charge-transfer excitations and the core-to- valence transitions that are probed with x-ray absorption spectroscopy (XAS). In this seminar, we will explore the hypothesis that these errors can be mitigated by choosing alternative initial reference orbitals for the excited-state calculation. By deviating from the usual ground-state molecular orbital reference, we aim to construct excited state methods that markedly enhance the accuracy of results compared to conventional TDDFT spectra for various excitation classes. We will take a journey through direct optimization methods such as State-Targeted Energy Projection (STEP) that attempt to optimize non-Aufbau solutions to the mean- field problem, to response theories like electron-affinity TDDFT (EA-TDDFT) that offer powerful solutions to the computational XAS problem by using core-ionized reference densities. Our exploration will take us from applying STEP to directly simulate the inception of hydrated electrons that are produced by charge-transfer-to- solvent excitations in I - (aq), to using EA-TDDFT alongside XAS to decipher the interplay of electronic and nuclear structure in liquids and prototypical photocatalysts.
Kevin Carter-Fenk
Dr. Kevin Carter-Fenk is an NIH Ruth L. Kirschstein Postdoctoral Fellow working with Professor Martin Head-Gordon at UC Berkeley on various topics related to the quantum chemistry of biological systems. Lately he has been working on new time- dependent density functional theory methods for the accurate and efficient calculation of X-ray absorption spectra. Another interest of Carter-Fenk’s is to make computationally tractable wave function approaches like many-body perturbation theory more useful in extended systems with a small band gap.
Hosted by Professor David Blank
Arkajit Mandal
Arkajit Mandal
Department of Chemistry
Columbia University
Abstract
Quantum Dynamics of Light and Matter between Two Mirrors
Placing molecules and materials between mirrors (or inside optical cavities) can enable the generation of exciting new chemical and physical phenomena in a highly controllable manner. In this talk, I will share a few of our theoretical works that demonstrate how photons can play the roles of a catalyst, a solvent, a quantum degree of freedom, beyond their usual role as a source of energy, to modify the chemical and physical properties of molecules and materials that are coupled to quantized radiation. First, I will discuss how quantum light-matter interactions can be utilized to modify the photochemistry of molecules and how a single photon can act as a catalyst to initiate multiple chemical reactions, leading to a quantum yield of more than one. Second, I will share our theoretical work that investigates how coupling molecular vibrations to vacuum radiation inside optical cavities can suppress or enhance the cleavage of specific chemical bonds, as seen in several recent experiments. I will share our theories that reveal that photons can act like solvents to enhance or suppress ground-state chemical kinetics. Next, I will show that quantum light- matter interactions can modify exciton-phonon interactions and can lead to the suppression of phonon-mediated decoherence. This enables fast ballistic transport in materials that are coupled to cavity radiation. I will share experimental results from our collaborators that confirm our theoretical predictions. Finally, I will share our theoretical framework, the quasi-diabatic propagation scheme, that allows for performing on-the-fly ab-initio quantum dynamics simulations, enabling us to investigate a wide range of problems, from photochemistry to polariton chemistry and beyond.
Arkajit Mandal
Dr. Arkajit is a postdoctoral research scientist at Columbia University, working with Prof. David R. Reichman. At Columbia, he is developing theoretical tools to understand and predict new physical effects that emerge when coupling material with a quantized radiation field inside an optical cavity. He is also actively collaborating with experimentalists at Columbia to realize these effects. Prior to this, Arkajit was a graduate student working with Prof. Pengfei Huo at the University of Rochester, where he obtained his Ph.D. in 2021. At Rochester, he developed new quantum dynamics methods and investigated new chemical reactivities enabled by coupling molecules to cavities.
Hosted by Professor Renee Frontiera
Professor Kade Head-Marsden
Professor Kade Head-Marsden
Department of Chemistry
Washington University in St. Louis
Abstract
Quantum information science as a playground for quantum chemists
There is an exponential cost to obtaining molecular electronic information pertinent to important processes such as catalysis, photosynthesis, and energy transfer. While quantum computing has shown early promise for reducing the scaling of these problems, practical realization of this promise remains challenging. The primary challenge arises due to the noise experienced by the quantum computer, limiting the size and complexity of possible algorithms and thus restricting the scope of possible applications. Detrimental noise occurs because qubits, the base unit of quantum computation, are fundamentally open quantum systems interacting with their environments. As we navigate this noisy quantum era, we need use-cases for the hardware we have access to now, along with creative paths forward. Here, I will discuss my group’s on- going efforts to utilize current quantum computers to simulate or emulate open quantum system dynamics, which are chemically relevant to processes such as molecular relaxation and excitonic energy transport, among others. I will also discuss an exploration of alternative computing platforms, based on molecular systems, and describe our efforts to model their electronic structure and potential as viable qubits.
Kade Head-Marsden
Professor Kade Head-Marsden is an assistant professor of chemistry at Washington University in St. Louis. She received her B.Sc. from McGill University in Mathematics and Chemistry, her Ph.D. from the University of Chicago in theoretical chemistry, and she was a postdoctoral fellow in the Paulson School of Engineering and Applied Sciences at Harvard University. Her research group focuses on developing methods to treat static and dynamic electronic structure using both classical and quantum mechanical computational resources.
Hosted by Professor Sapna Saupria
Jan-Niklas Boyn
Jan-Niklas Boyn
Department Mechanical & Aerospace Engineering
Princeton University
Abstract
From Reduced Density Matrix Methods to Multi-Level Embedding: Elucidating Fundamental Interactions Governing Quantum Materials and Carbon Mineralization
For the precise and tractable computation of molecular electronic properties and processes, it is essential to develop quantum mechanical methods that include electron correlation, as well as extended structural and dynamic effects at affordable computational cost. In the first part of this seminar, I will review recent advances in the development of reduced-density-matrix (RDM) based theories, which enable accurate computation of electronic structure without explicit consideration of an electronic wavefunction, offering lower scaling alternatives to wavefunction-based theories. I will demonstrate their ability to elucidate fundamental interactions from which novel electronic and magnetic properties emerge in multi-metallic open-shell transition metal complexes. In the second part of the seminar, I will discuss how multi-level quantum embedding simulations, harnessing both correlated wavefunction theories and molecular dynamics, may be utilized to describe dynamic processes occurring in solution. Focus will lie on the atomic-level processes governing carbonate mineralization, a promising strategy for carbon capture and storage.
Jan-Niklas Boyn
Dr. Jan-Niklas Boyn is a postdoctoral research associate in Prof. Emily Carter’s group in the Department of Mechanical and Aerospace Engineering at Princeton University. His research utilizes multi-level embedding simulations to elucidate atomic- level processes pertaining to carbon capture. He received a MChem from the University of Oxford, where he carried out research in computational organic chemistry under Prof. Robert Paton, and a Ph.D. in theoretical chemistry from the University of Chicago in Prof. David Mazziotti’s group, developing reduced density matrix based electronic structure methods.
Hosted by Professor Sapna Sarupria
Professor Xavier Roy
Professor Xavier Roy
Department of Chemistry
Columbia University
Abstract
Next level quantum materials
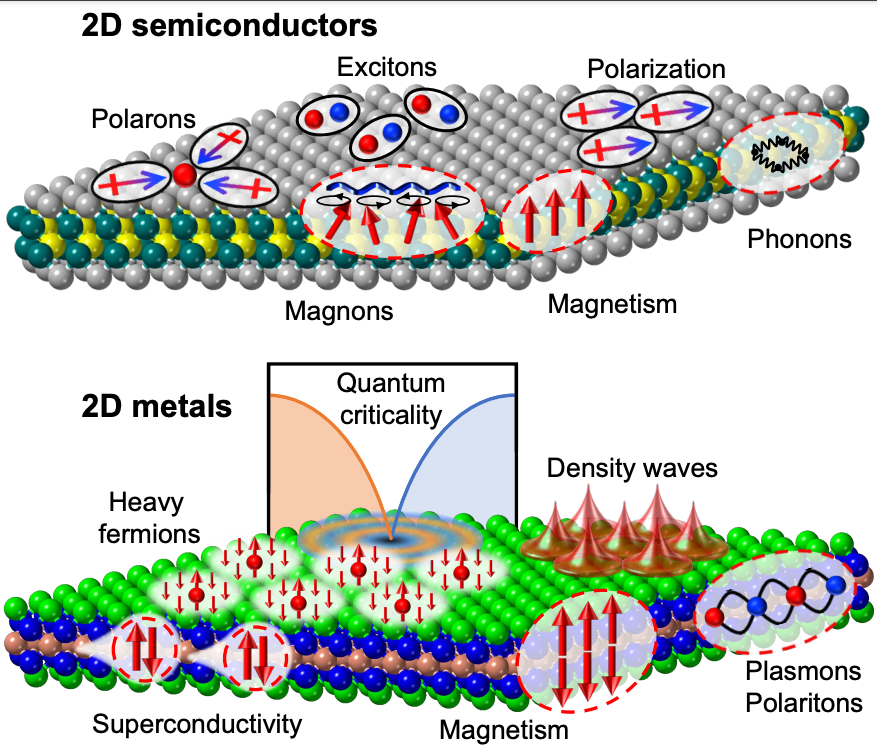
Over the past 15 years, two-dimensional (2D) materials have garnered significant attention due to their exceptional physical, mechanical, and chemical properties, as well as our ability to seamlessly incorporate them into various devices. In this seminar, I will present our recent advancements in the development of the next generation of 2D materials. To begin, I will discuss our efforts to assemble atomically defined clusters of atoms, which we refer to as “superatoms,” into hierarchical 2D structures. These superatoms mimic the role of atoms in conventional 2D “atomic” solids, resulting in remarkable material properties. I will highlight recent synthetic breakthroughs and explore the distinctive transport behaviors that emerge due to the atomic precision of the 2D lattice and the specific interactions between these building blocks. In the second part of the presentation, I will introduce novel atomic 2D materials that have been developed in our laboratories in recent years. Specifically, I will delve into how magnetic order strongly influences optical transitions in the 2D magnetic semiconductor CrSBr. Additionally, I will provide insights into the synthesis and characterization of CeSiI, the first f-electron-based heavy fermion metal, which also falls under the category of 2D van der Waals (vdW) materials. Conceptually, our synthetic design takes a traditional 3D intermetallic heavy fermion compound and slices it into atomically thin vdW sheets by incorporating iodine into the structure. The resulting material is cleavable and effectively 2D electronically, even in bulk crystals.
Xavier Roy
Professor Xavier Roy received a B.Eng. (2002) and a Master of Applied Science (2005) in Chemical Engineering from Ecole Polytechnique of Montreal, performing research under the guidance of Prof. Basil Favis. He earned his Ph.D. in Chemistry with Prof. Mark MacLachlan at the University of British Columbia in 2011, working as an NSERC Alexander Graham Bell Scholar. He went on to do postdoctoral research as a Canada NSERC Postdoctoral Fellow with Prof. Colin Nuckolls at Columbia University from 2011 to 2013. He joined the Columbia University Faculty in 2013 as an Assistant Professor of Chemistry, and was promoted in 2018 to the rank of Associate Professor and received tenure in 2020.
Hosted by Professor Gwendolyn Bailey
Laura McCaslin
Laura McCaslin
Sandia National Laboratories
Abstract
A Bottom-Up Approach to Rational Design of Molecular Materials
Organic donor-acceptor (D-A) co-crystals are a promising class of molecular materials for next-generation optoelectronic devices. High-throughput screening and machine learning methods employ structure-function relationships to predict combinations of donor and acceptor molecules with desired properties. However, optical properties of materials are particularly difficult to predict due to the computational expense of performing electronically excited state calculations. Furthermore, calculations of a molecular material’s radiative and non-radiative lifetimes cannot be straightforwardly computed from D-A cluster models, as orbital energy differences, couplings, and oscillator strengths differ substantially between the molecular dimer and a crystalline material. In this talk I present a novel approach to predicting radiative and non-radiative lifetimes in D-A co-crystals based on orbital structure-function relationships. The basis for this approach is the striking similarity of orbital character between the D-A dimer and D-A co- crystal, especially in the ground and first excited states. Furthermore, these orbital characteristics are related to the degree of charge transfer in different electronic states, which are key quantities for predicting radiative and non-radiative lifetimes. Here I present a novel metric for predicting the degree of charge transfer in the S1 (HOMO → LUMO) electronic state using ground state orbital analysis alone, enabling low cost predictions of radiative and non-radiative lifetimes in D-A co-crystals for future high-throughput screening approaches. We use this metric to correlate S1 charge transfer with internal conversion and intersystem crossing timescales in D-A co-crystals, measured with UV pump, UV-Vis probe ultrafast transient absorption spectroscopy (TAS). Non-adiabatic mixed quantum-classical dynamics (NA-MQC) calculations of D-A dimer models enable characterization of the ultrafast TAS signatures to unravel the complex physics underlying long exciton lifetimes in D-A co-crystals.
Laura McCaslin
Dr. Laura McCaslin is a staff scientist in theoretical chemical physics at Sandia National Laboratories in Livermore, CA. Her research program specializes in studying the electronic structure, photochemistry, and dynamics of molecular systems. During her undergraduate studies at the University of California Berkeley, McCaslin performed research under the advisement of Prof. Martin Head- Gordon on the electronic structure of microsolvated ions. After completing a BS in Chemical Biology in Spring 2012, she began her PhD studies at the University of Texas Austin in Fall 2012. McCaslin performed research on the electronic and vibrational structure of small molecules under the advisement of Prof. John Stanton, completing her PhD in Fall 2016. She began a postdoc position at the Hebrew University of Jerusalem in January 2017 under the advisement of Prof. R. Benny Gerber, where she employed molecular dynamics calculations to study reactions at atmospheric aerosol interfaces. She began her independent research program at Sandia National Labs in Spring 2020, where her research in electronic structure and molecular dynamics has spanned many fields, including atmospheric chemistry, materials discovery and characterization, quantum control, and photodynamics.
Hosted by Professor Ilja Siepmann
Professor Alexandra Velian
Professor Alexandra Velian
Department of Chemistry
University of Washington
Abstract
Molecular Catalysts Go Nano: Installing Active Sites on Clusters and Nanosheets
A central research goal in the Velian group is to create next-generation single atom catalysts poised to harness the cooperativity between the active site and chemically non-innocent supports. To emulate defect sites in inorganic heterogeneous catalysts in a controlled fashion, we embed well-defined active sites on the surface of clusters and 2D nanosheets.
The first part of this seminar will introduce a class of atomically precise nanoclusters distinguished by the presence of discrete active sites that engage dynamically with the non-innocent metal chalcogenide cluster support, and with substrates. Together with their scalable syntheses, solution processability and their ease of characterization using molecular methods, this family of nanoclusters provide atom level insights into the metal/support interface, and how the ensuing cooperativity can be harnessed to dramatically alter catalytic activity.
The second part of the talk will introduce an orthogonal approach to catalyst engineering, in which the basic principles of organometallic- phosphine chemistry are harnessed to anchor single site active sites on the surface of two-dimensional black phosphorus nanosheets. Molecular synthetic and characterization strategies are leveraged to probe the bonding and functionality of the modified nanosheets.
Alexandra Velian
Professor Velian began her independent career at the University of Washington in 2017. A central goal in the group is to create next-generation catalysts geared to turn green- house gases like methane and carbon dioxide into value added products. Her approach is to use molecular strategies to synthesize single-site catalysts that harness metal-support interactions, and shine light on processes that govern the substrate/active sites/ support interactions.
Professor Velian’s scientific and academic contributions have been recognized with several awards and distinctions, including the Inorganic Chemistry Lectureship Award (2023), the Camille Dreyfus Teacher Scholar Award (2023), the Marion Milligan Mason AAAS Award (2023), the C&EN Talented 12 distinction (2022), a Cottrell Scholar Fellowship (2020), the NSF Career Award (2019), the Young Investigator Award – ACS Division of Inorganic Chemistry (2016) and the Alan Davison Prize for the Best Thesis in Inorganic Chemistry at MIT (2015). Professor Velian completed her undergraduate studies in chemistry at Caltech, where she conducted research primarily with Professor Theodor Agapie. As the first member of his group, she developed the synthesis of low-valent mono- and bimetallic complexes supported by a terphenyl diphosphine framework.
She received her Ph.D. under the direction of Professor Christopher C. Cummins at MIT, where she developed the synthesis of anthracene and niobium- supported precursors to reactive phosphorus fragments and studied their behavior using chemical, spectroscopic, and computational methods. Notably, this work gave rise to the synthesis of the 6π all-inorganic aromatic anion heterocycle P2N3−, produced in the “click” reaction of P2 with the azide ion.
Following her PhD, Alexandra was a Materials Research Science & Engineering Center postdoctoral fellow with Professor Colin Nuckolls at Columbia University, where she worked on creating well-defined functional nanostructures by linking atomically precise metal chalcogenide clusters.
Professor Velian was born in Tulcea, Romania, and currently lives in Seattle with her husband, daughter and two cats. Outside chemistry, she loves the outdoors and ballroom dancing.
Hosted by Professor Gwendolyn Bailey
Professor Kathryn Riley
Professor Kathryn Riley
Department of Chemistry
Swarthmore College
Abstract
Embodying DEI in Teaching, Research, and Outreach
In this talk, I will discuss the challenges and triumphs in my journey as a black woman in chemistry. I will also share how my personal experiences have shaped my current approaches to DEI work. Specifically, I will outline how I have designed my courses for inclusivity, how I have built and sustained a diverse research team, how my research enables the participation of diverse scholars in nanomaterials research, and the work I am doing to improve K-12 education in underserved communities.
Kathryn Riley
Dr. Kathryn Riley is an Assistant Professor in the Department of Chemistry and Biochemistry at Swarthmore College. She received her Ph.D. from Wake Forest University in 2014 and was a National Research Council (NRC) postdoctoral fellow at the National Institute of Standards and Technology (NIST) from 2015-2016. Before her current appointment, she was a Consortium for Faculty Diversity (CFD) postdoctoral fellow at Swarthmore from 2016-2018. Dr. Riley’s research involves the development of analytical techniques for the characterization of nanomaterials (NMs) and their dynamic physical and chemical transformations in biological and environmental matrices. Her research group specifically aims to broaden participation in the field by developing techniques that provide new quantitative insights in less time and at a reduced cost when compared to more commonly employed methods. Projects in her group span the analysis of engineered NMs (metal and metal oxide NMs, DNA nanostructures) and incidental NMs (nano and microplastics).
Hosted by Alexander Umanzor
Professor Kathryn Riley
Professor Kathryn Riley
Department of Chemistry
Swarthmore College
Abstract
Chemistry in Context: Re-envisioning Introductory Chemistry to Serve DEI Goals
In the Fall of 2022, the introductory chemistry course at Swarthmore (Chem 010) underwent significant revision to address several curricular priorities, including improved accessibility, engagement, and persistence for learners from diverse backgrounds. In this talk, I will discuss the process through which Swarthmore chemistry faculty revised Chem 010 to its present form. I will begin by discussing the national landscape of chemistry education, its relation to the previous version of Chem 010, and its role in motivating our curricular changes. Then, I will discuss the departmental and course level planning needed to reform our curriculum. Finally, I will present our new model for the course and outcomes from our first semester of implementation.
Kathryn Riley
Dr. Kathryn Riley is an Assistant Professor in the Department of Chemistry and Biochemistry at Swarthmore College. She received her Ph.D. from Wake Forest University in 2014 and was a National Research Council (NRC) postdoctoral fellow at the National Institute of Standards and Technology (NIST) from 2015-2016. Before her current appointment, she was a Consortium for Faculty Diversity (CFD) postdoctoral fellow at Swarthmore from 2016-2018. Dr. Riley’s research involves the development of analytical techniques for the characterization of nanomaterials (NMs) and their dynamic physical and chemical transformations in biological and environmental matrices. Her research group specifically aims to broaden participation in the field by developing techniques that provide new quantitative insights in less time and at a reduced cost when compared to more commonly employed methods. Projects in her group span the analysis of engineered NMs (metal and metal oxide NMs, DNA nanostructures) and incidental NMs (nano and microplastics).
Hosted by Alexander Umanzor