Most precise measurements of sickle cell disease building blocks could lead to new treatments
Research could help 20 million people worldwide who suffer with sickle cell disease
MINNEAPOLIS / ST. PAUL (03/28/2019) — In a breakthrough study of sickle cell disease, biomedical engineers in the University of Minnesota College of Science and Engineering have revealed that the building blocks of the disease are much less efficient at organizing than previously thought.
The findings open the door to new treatments, including new medicines that could be prescribed at lower doses, for the approximately 20 million people worldwide who suffer from the lifelong disease.
The study, which includes the most precise measurements ever of the disease at the molecular level, is published in Science Advances, a journal of the American Association for the Advancement of Science.
“Even though it has been known for decades what causes sickle cell disease at the molecular level, no one has ever studied the disease at this level of detail,” said David Wood, an associate professor of biomedical engineering at the University of Minnesota and a lead author of the study. “What we found at the nanoscale was quite surprising. We found that the disease self-assembly process is less efficient than we thought, which means that it could be easier to develop new medicines that would be effective at lower doses and would cause fewer side effects for patients.”
Sickle cell disease is an inherited lifelong disorder that causes problems in the protein within red blood cells, called hemoglobin. The hemoglobin molecules carry oxygen throughout the body. With sickle cell disease, hemoglobin molecules form into fibers that act like stiff rods within the red blood cells. The formation of these fibers stiffens the red blood cells and can change the shape from disc-shaped to crescent, or sickle, shape.
When the red blood cells stiffen, they contribute to blockages in blood vessels that slow or stop the flow of blood. When this happens, oxygen can’t reach nearby tissues. The lack of oxygen can affect the entire body causing severe pain, increasing the risk of strokes, and causing infections.
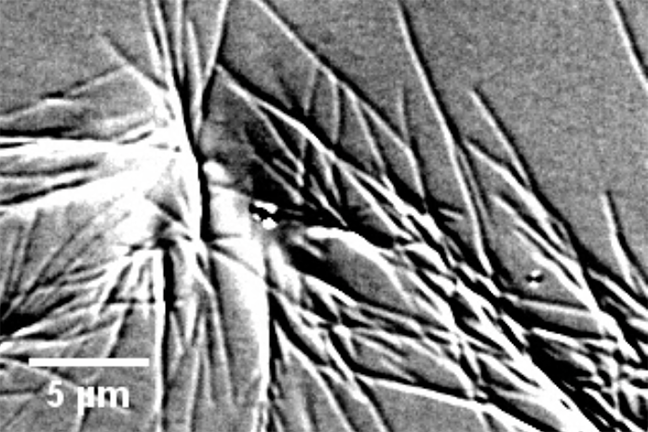
The University of Minnesota researchers in this study performed the highest-ever resolution measurements of single sickle hemoglobin fiber assembly in action using microscopes and cameras that can measure the molecules at the nanoscale. Their measurements show that the rates of sickle hemoglobin addition and loss have been underestimated in previous studies. The new results reveal that the sickle hemoglobin self-assembly process is very rapid and inefficient. They found that the process is 4 percent efficient versus 96 percent efficient as researchers previously thought.
“It’s kind of like building a tower by stacking LEGOs,” Wood said. “For every 100 LEGOs only four actually stay on as part of the tower. I would have to stack another 100 LEGOs to get another four to stay on. This shows us that this is a much more inefficient process than previously thought and that it wouldn’t take as much medicine to disrupt this process.”
Currently only two FDA-approved medicines are available to those with sickle cell disease. While newer alternatives to existing medications are now in development to treat the disease, including stem cell transplants and gene therapy, Wood said those treatments will probably not be available to the millions of people in the developing world who suffer from the disease.
Globally, it is estimated that 300,000 infants are born annually with sickle cell disease. In the developing world, it remains a major killer of infants and children, particularly in sub-Saharan Africa and India, where an estimated 50 to 90 percent of infants born with sickle cell disease will die before age 5. In the United States, nearly 100,000 individuals have sickle cell disease. The Centers for Disease Control and Prevention estimates that sickle cell disease affects 1 out of every 365 black or African-American births, and 1 out of every 16,300 Hispanic-American births. The U.S. median life expectancy for those with sickle cell disease is 47 years.
“We are hoping the new information we revealed in this study could make a big impact worldwide to develop medicines and other treatments that help millions of people,” Wood said.
Wood collaborated with fellow University of Minnesota Department of Biomedical Engineering Professor David Odde and post-doctoral researcher Brian Castle on this study. The research was built upon Odde’s previous work studying a similar assembly of microtubules in cancer.
“We thought we could apply what we learned studying microtubules, which are important targets for cancer treatment, to sickle cell disease, and we were right,” Odde said. “This shows the power of collaborative research. My area of expertise was studying microtubule self-assembly in cancer, and Professor Wood has been studying sickle cell disease for more than a decade. By teaming up, and bringing in Dr. Castle’s expertise on nanoscale imaging and analysis, we feel we have made a major breakthrough.”
This research was funded primarily by the National Institutes of Health grants R01GM76177 and R01HL132906.
To read the entire study entitled “Rapid and inefficient kinetics of sickle hemoglobin fiber growth,” visit the Science Advances website.